Direct Photocatalytic Synthesis of Acetic Acid from Methane and CO at Ambient Temperature Using Water as Oxidant
- Type: Journal Article
- Author: Dong, Chunyang; Marinova, Maya; Tayeb, Karima Ben; Safonova, Olga V.; Zhou, Yong; Hu, Di; Chernyak, Sergei; Corda, Massimo; Zaffran, Jérémie; Khodakov, Andrei Y.; Ordomsky, Vitaly V.
- Journal: Journal of the American Chemical Society
- Volume: 145
- Issue: 2
- Pages: 1185-1193
- Year: 2023
- DOI: 10.1021/jacs.2c10840
Abstract
Direct functionalization of methane selectively to value-added chemicals is still one of the main challenges in modern science. Acetic acid is an important industrial chemical produced nowadays by expensive and environmentally unfriendly carbonylation of methanol using homogeneous catalysts. Here, we report a new photocatalytic reaction route to synthesize acetic acid from CH4 and CO at room temperature using water as the sole external oxygen source. The optimized photocatalyst consists of a TiO2 support and ammonium phosphotungstic polyoxometalate (NPW) clusters anchored with isolated Pt single atoms (Pt1). It enables a stable synthesis of 5.7 mmol·L–1 acetic acid solution in 60 h with the selectivity over 90% and 66% to acetic acid on liquid-phase and carbon basis, respectively, with the production of 99 mol of acetic acid per mol of Pt. Combined isotopic and in situ spectroscopy investigation suggests that synthesis of acetic acid proceeds via a photocatalytic oxidative carbonylation of methane over the Pt1 sites, with the methane activation facilitated by water-derived hydroxyl radicals.
Files and Links
- Url: https://doi.org/10.1021/jacs.2c10840
- Local Library: Zotero
Detail
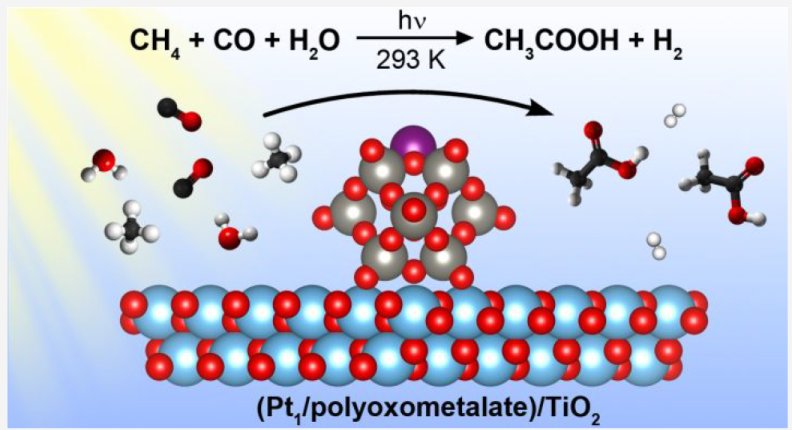
Key points:
-
Photocatalytic reaction at room temperature, water as sole external oxygen source
-
TiO2 support, Pt single atom achored by NPW (ammonium phosphotungstic polyoxometalate)
-
5.7 mmol/L acetic acid in 60 h (sel>90% in liquid phase, 66% in carbon basis), TON of 99
-
Isotopic and in situ spectroscopy investigation
-
Photocatalytic oxidative carbonylation over Pt, methane activation facilitated by water-derived hydroxyl radicals
Learning points:
-
IR with temperature-programmed desorption confirmed atomically dispersion of Pt
-
The investigation of mechanism is impresive (especially EPR and XANES)
-
The acidity drives the Pt/NPW aggregates’ disassembly over the basic TiO2 surface.
-
The variation of oxidation state of W species is interesting
-
The synergy of Pt/NPW and TiO2 is surprising
-
Mars van Krevelen type mechanism
Role of Water
Sushkevish et al.: soft oxidant and intermediate stabilizer in anaerobic oxidation of methane for methanol
Liu et al.: O-provider and site-blocker in methane oxidaiton to methanol
Photocatalytic Synthesis
4 h (2 bar of CO, 8 bar of CH4), different catalysts
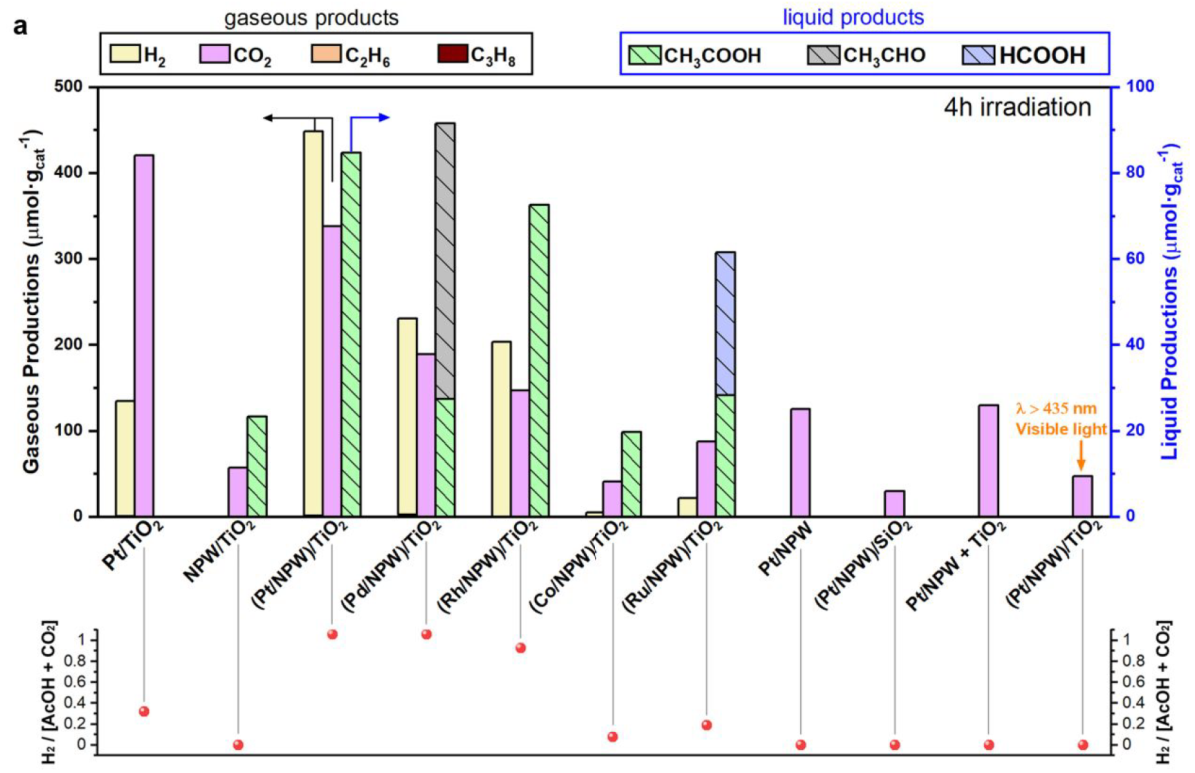 Pt/TiO2: a part of the oxygen species for CO and/or methane oxidation to CO2 comes from the catalysts
Pt/TiO2: a part of the oxygen species for CO and/or methane oxidation to CO2 comes from the catalysts
The production of AcOH was observed only for (M/ NPW)/TiO2 (M = Pt, Pd, Rh, Co, and Ru) and NPW/TiO2
No oxygen was detected during the CH4 photocatalytic conversion.
(Pt/NPW)/TiO2 irradiated with visible light: AcOH synthesis over (Pt/NPW)/TiO2 is essentially driven by photoinduced charge carriers from TiO2
lower hydrogen production for other catalysts can be attributed to the partial reduction of NPW and metal oxide species with H2, which do not contain Pt, Pd, and Rh.
12 h, NPW/TiO2 VS (Pt/NPW)/TiO2
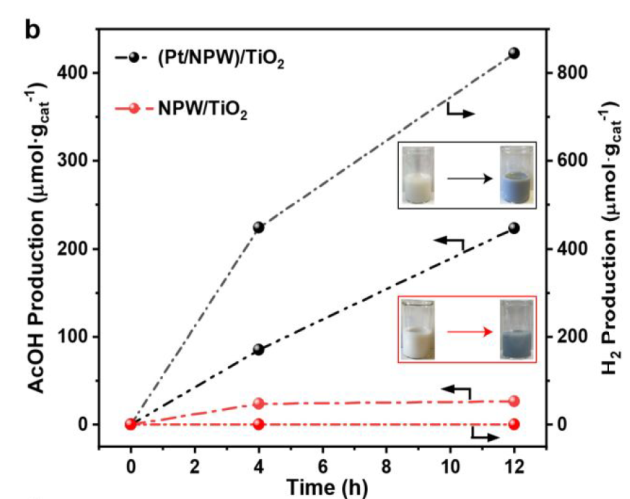 catalysts changed color to dark blue: formation of partially reduced NPW
catalysts changed color to dark blue: formation of partially reduced NPW
Pt species are capable of utilizing H2O as the external oxidant for regeneration of NPW oxygen species and simultaneously produce H2.
Ratio of CO/CH4
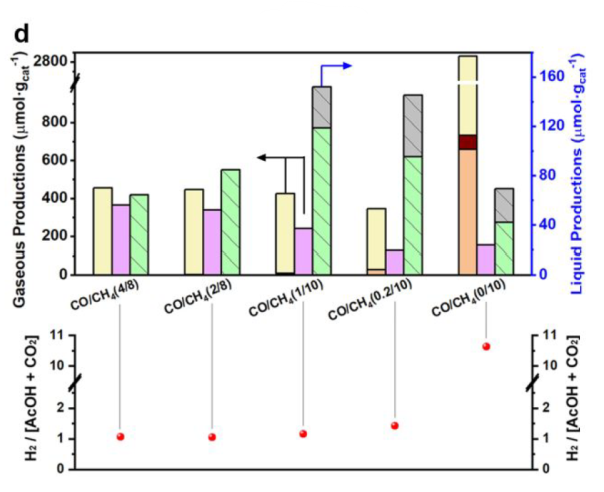
Acetaldehyde: derived from the intermediates
CO2 and H2 were produced by WGS reaction
Best condition: 1 bar of CO, 10 bar of CH4, 15 h
Composition of (Pt/NPW)/TiO2
Varying the content of Pt/NPW in the catalyst and Pt content in NPW
Best composition: 0.2 wt % Pt loading and the weight ratio between Pt/NPW and TiO2 at 3:10
Catalyst Stability
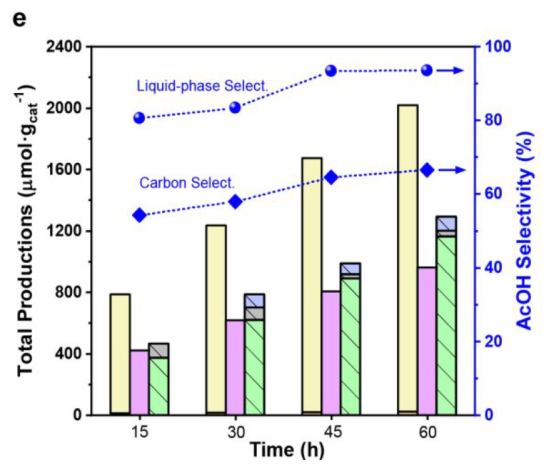
CO adsorption over Pt sites during the reaction preserves the AcOH from rapid decomposition (Figure S10)
Catalyst Characterization
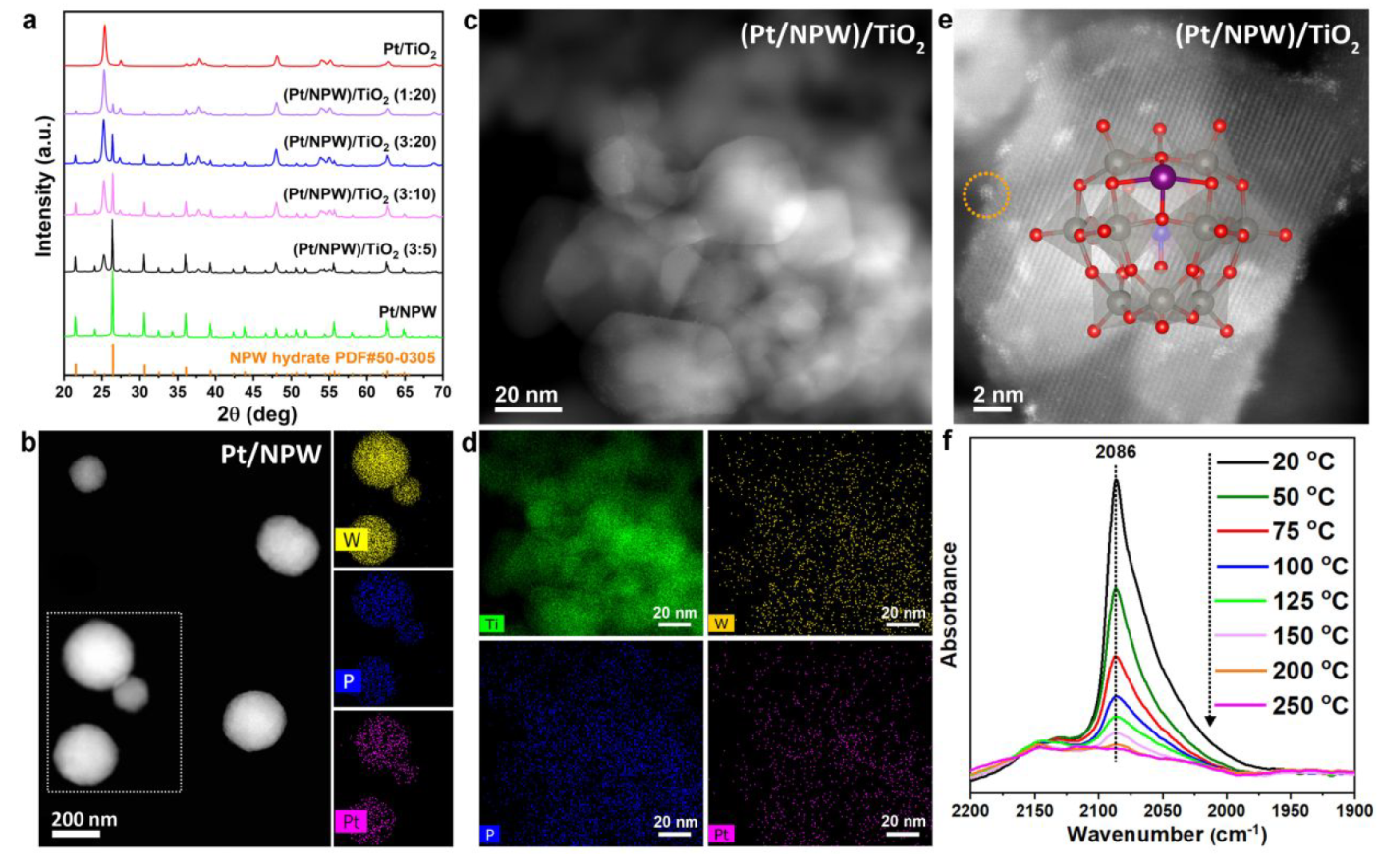
a. XRD patterns, b. HAADF-STEM and EDS-mapping of Pt/NPW, c. HAADF-STEM of (Pt/NPW)/TiO2, d. EDS-mapping of (Pt/NPW)/TiO2, e. Atomic-resolved HAADF-STEM of (Pt/NPW)/TiO2, f. IR spectra of temperature-programmed CO desorption over (Pt/NPW)/TiO2
Atomic dispersion of primary Pt species over the catalyst was confirmed by IR of TPD.
The acidity drives the Pt/NPW aggregates’ disassembly over the basic TiO2 surface.
Mechanism
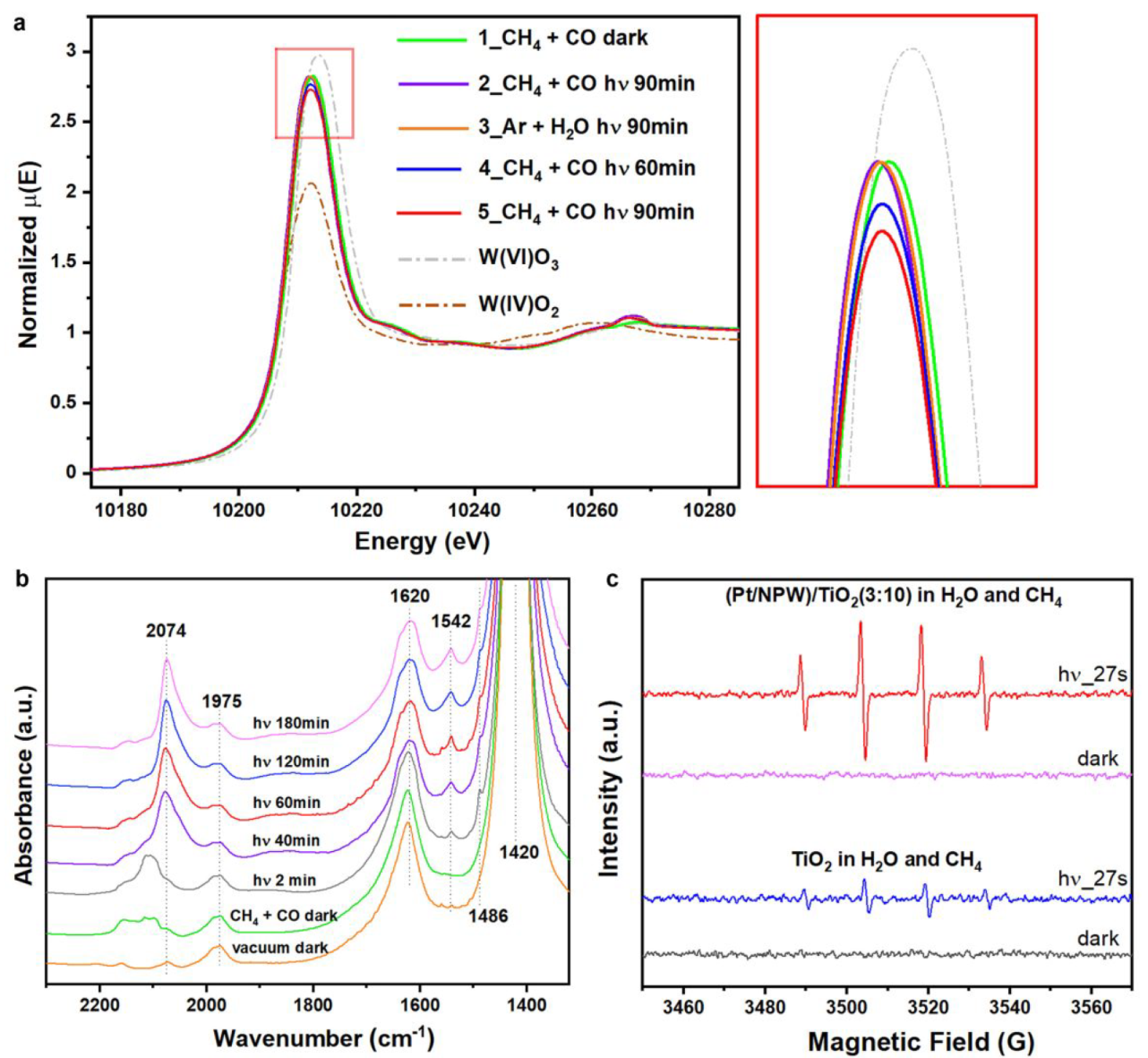
a. In situ W L3-edge XANES spectra of (Pt/NPW)/TiO2(3:20) b. In situ IR spectra of (Pt/NPW)/TiO2(3:5) c. In situ EPR spectra with DMPO radical-trapping agent
Gradual reduction of W species was observed.
IR absorption bad: NH4+ at 1420, absorbed H2O at 1620, *CH3COO at 1486 and 1542, absorbed CO on cationic Pt between 2159 and 2100, CO on reduced Pt at 2074
Pt/NPW clusters promote the charge separation efficiency of TiO2 by accepting photoexcited electrons (Figure S19)
·OH played fundamental roles in promoting methane activation
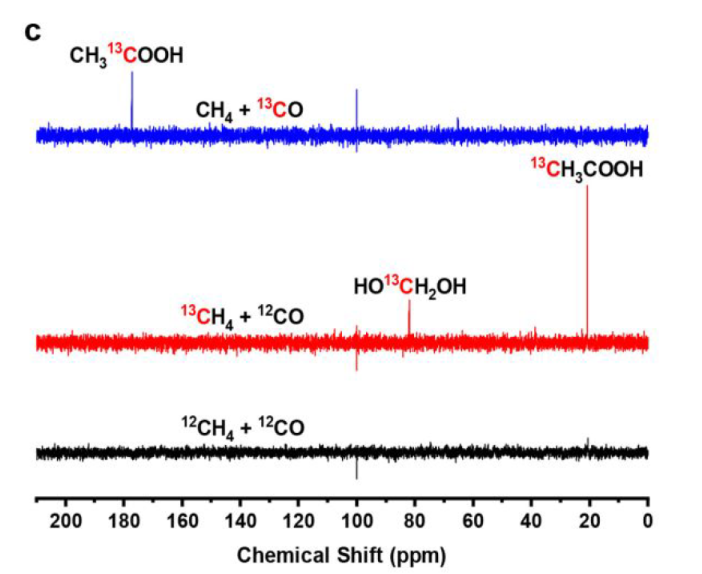
The methyl groups are derived from CH4, while acyl groups of AcOH are produced from CO.
CO2 is principally produced from CO by the photocatalytic WGS reaction (Figure S18)
Proposed Mechanism:
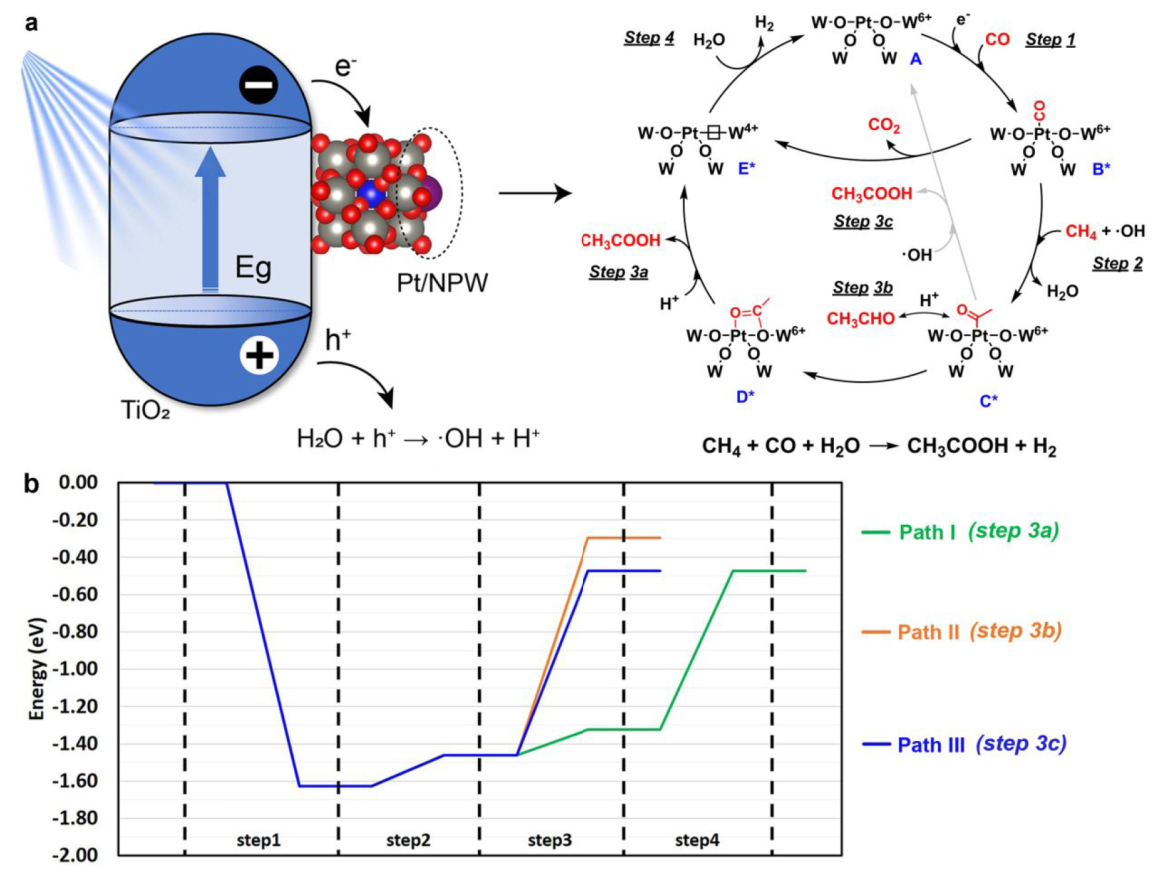
Path Ⅲ is more favored (Mars van Krevelen type mechanism)